


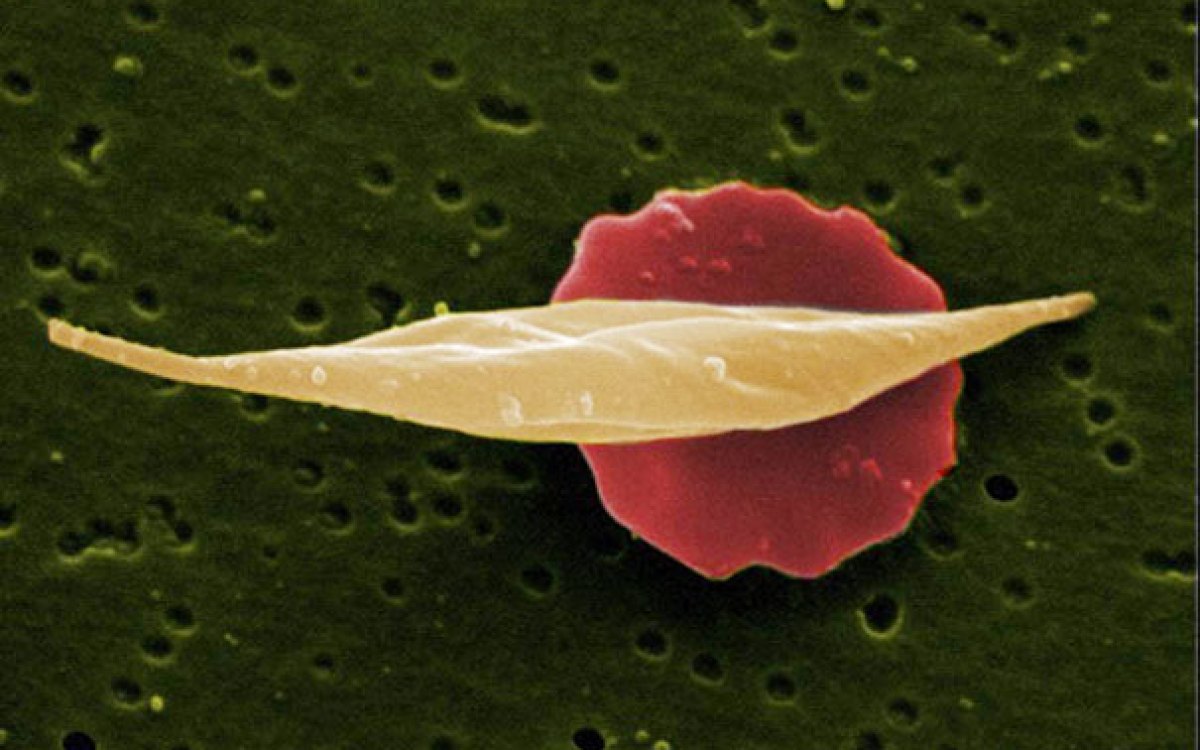
Sickle cell disease is caused by a mutation in hemoglobin, the oxygen-carrying protein in red blood cells, which causes the usually pliant cells to stiffen and assume a curved or “sickle” shape.
Courtesy of Wikimedia Commons
Health
Gene therapy for sickle cell disease passes key preclinical test
Decades-old discovery about fetal hemoglobin is on track for clinical trial in the coming year
More like this
A precision-engineered gene therapy virus, inserted into blood stem cells that are then transplanted, markedly reduced sickle-induced red-cell damage in mice with sickle cell disease, researchers from Dana-Farber/Boston Children’s Cancer and Blood Disorders Center report today in the Journal of Clinical Investigation.
The work sets the stage for bringing a decades-old discovery about sickle cell disease to the bedside. A clinical gene therapy trial, using a virus rendered harmless in the laboratory, is expected to launch in the coming year.
Sickle cell disease is caused by a mutation in hemoglobin, the oxygen-carrying protein in red blood cells, which causes the usually pliant cells to stiffen and assume a curved or “sickle” shape. These sickle cells live for less time than those with healthy hemoglobin, leading to anemia. They also bind together, leading to blood-flow blockage that can cause severe pain and organ damage throughout the body.
The new gene therapy is the culmination of research going back to the 1980s that found that sickle cell disease is milder in people whose red blood cells carry a fetal form of hemoglobin. Fetal hemoglobin normally tails off after birth, but in 2008, Dana-Farber/Boston Children’s researchers Stuart Orkin and Vijay Sankaran showed that suppressing a gene called BCL11A — which acts as an “off” switch — could restart fetal hemoglobin production. In 2011, using this approach, they corrected sickle cell disease in mice, replacing much of the defective beta (“adult”) hemoglobin that causes sickling with healthy fetal hemoglobin.
To turn this insight into a therapy, a team led by Dana-Farber/Boston Children’s researchers David A. Williams, Christian Brendel, and Swaroopa Guda faced a major problem: BCL11A, they discovered, has an important role in blood stem cells. When it is silenced in mice with the sickle cell mutation, blood stem cells cannot engraft long-term in the animals’ bone marrow and eventually become depleted. This failure to engraft could render gene therapy ineffective and also cause serious problems with blood development in general.
To circumvent this, the team performed some intricate engineering, creating a gene therapy virus that silences BCL11A selectively — only in precursors of red blood cells. Blood stem cells treated with this gene therapy, transplanted into the mice, engrafted successfully and reduced signs of sickle cell disease (hemolytic anemia and increased numbers of reticulocytes).
Moreover, in red blood cells from the mice and from four patients with sickle cell disease, fetal hemoglobin crowded out the sickling beta hemoglobin, making up at least 80 percent of the total hemoglobin in the cell. This exceeded the researchers’ original goal of 50 percent fetal hemoglobin — likely more than enough to avoid clinical sickle cell disease.
“The tendency for a red blood cell to sickle is proportional to how much nonsickling versus sickling hemoglobin it has,” explained Williams, the study’s senior author and president of Dana-Farber/Boston Children’s. “BCL11A represses fetal hemoglobin, which does not lead to sickling, and also activates beta hemoglobin, which is affected by the sickle-cell mutation. So when you knock BCL11A down, you simultaneously increase fetal hemoglobin and repress sickling hemoglobin, which is why we think this is the best approach to gene therapy in sickle cell disease.”
Precision engineering
Selectively knocking down BCL11A involved several layers of engineering. As the core of their gene therapy vector, the researchers used a snippet of genetic code called a short hairpin RNA (shRNA) that inactivates the BCL11A gene. To get it into cells, they embedded the shRNA in another bit of code, a microRNA, that cells generally recognize and process. To make this assembly work in the right place at the right time, they hooked it to a promoter of beta hemoglobin expression, together with regulatory elements active only in precursors of red blood cells. Finally, they inserted the whole package into a lentivirus engineered for safe use in humans.
Blood stem cells from mice and the sickle cell patients were then exposed to the manipulated virus, taking up the new genetic material. The genetically engineered red-blood-cell precursors began producing fetal hemoglobin rather than the mutated beta hemoglobin, just as intended.
A double benefit
Other gene therapy trials for sickle cell disease are underway or being developed. Rather than interfering with BCL11A, these approaches are introducing genes that encode fetal hemoglobin itself or a corrected beta hemoglobin that doesn’t sickle.
Williams believes the BCL11A-based approach to gene therapy for sickle cell disease will substantially increase the ratio of nonsickling versus sickling hemoglobin. He notes that this approach may also be beneficial in beta-thalassemia, another blood disorder involving defective beta hemoglobin.
Williams has launched multiple other gene-therapy trials at Dana-Farber/Boston Children’s, which has become a world-leading hub of gene therapy for pediatric genetic diseases. His team is now taking the final steps toward FDA clearance for a clinical gene-therapy trial in sickle cell disease that is expected to begin in early 2017.
Brendel and Guda are co-first authors on the paper. The work was supported by the Clinical and Translational Investigation Program at Boston Children’s Hospital, the National Institutes of Health, the Doris Duke Charitable Foundation, and sponsored research support from Bluebird Bio.





