


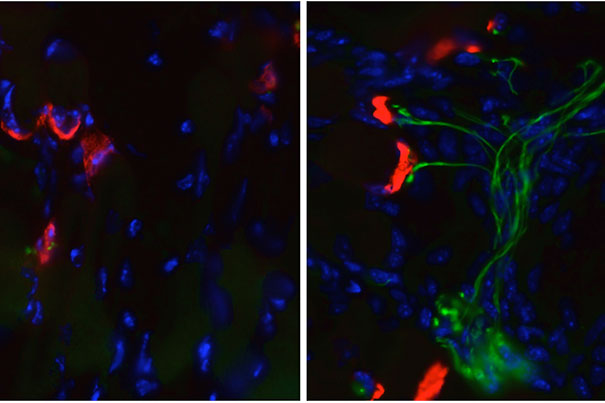
In the right-hand picture, an SMA mouse treated with guanabenz, a compound known to reduce cellular stress, shows healthy junctions between muscle cells (red) and motor nerve fibers (green). In contrast, the untreated SMA mouse (left) has lost its nerve fibers, leaving the muscle cells with no way to receive signals from neurons in the spinal cord.
Photo courtesy of Shi-Yan Ng
Health
Hope against disease targeting children
Researchers identify molecular changes in genetic ailment, see ties to ALS behavior
Harvard Stem Cell Institute (HSCI) researchers studying spinal muscular atrophy (SMA) have found what they term “surprising similarities” between this childhood disorder that attacks motor neurons and amyotrophic lateral sclerosis (ALS), more commonly known as Lou Gehrig’s disease.
The findings have been published online by the journal Cell Stem Cell.
The research team led by HSCI principal faculty member Lee Rubin uncovered molecular changes that explain, at least in part, why motor neurons rather than others are affected by the illness. Unlike ALS and other neurodegenerative diseases, which tend to manifest later in life, SMA strikes infants. Unlike ALS, SMA is a genetic disorder that causes a range of outcomes, with the milder form leaving some children confined to wheelchairs, and the more severe form causing paralysis and death before the second birthday.
Though not as well-known as ALS, SMA is “the most frequent fatal genetic disease of young children,” said Rubin, a professor in Harvard’s Department of Stem Cell and Regenerative Biology (HSCRB). About one in 50 people are carriers of SMA and about one in 5,000 children are born with the disease.
Rubin said researchers are still working to determine the mechanisms of SMA. “It has never been clear why motor neurons — which relay signals from the brain to the muscles via the spinal column — die selectively,” Rubin said. “It is clear motor neurons die well before other kinds of cells, even other kinds of spinal-cord neurons, and the mystery has been trying to understand that.”
A research team composed of HSCI investigators at the Broad Institute of Harvard and MIT and HSCRB, including HSCI principal faculty member John Rinn, worked in collaboration with the SMA Foundation’s Pediatric Neuromuscular Clinical Research Network and Wendy Chung from Columbia University College of Physicians and Surgeons to make neurons from donors with SMA of varying degrees of severity.
The researchers first determined that the neurons in a dish behaved similarly to the way neurons would behave in an SMA patient. Not only did motor neurons die before other types of neurons, but motor neurons derived from patients with severe SMA died very quickly in comparison to those made from patients with a milder form of the disease.
Then, using a method of intracellular cell labeling developed in HSCI co-director Doug Melton’s lab, the researchers separated motor neurons from other types of neurons in the dish, carried out an RNA sequencing analysis, and compared SMA motor neurons to those from healthy individuals.
Healthy cells have mechanisms that help maintain an appropriate quantity of protein made by genes that are turned on within a given cell. There is a balance between protein production and protein degradation. Over time, neural cells targeted by late-onset neurodegenerative diseases — Parkinson’s, Alzheimer’s, Huntington’s, even ALS — lose their ability to maintain that balance. Proteins build up, and the clutter stresses the cell. If cells cannot resolve the issue, they shut down and die. After a number of cells have died, patients start to experience the effects of their disease.
SMA, however, appears to work in the opposite way. There isn’t too much but too little of a specific protein, called survival of motor neuron (SMN), because the gene that codes that protein is broken. The researchers learned that a reduction in this protein affects the cells’ ability to process other proteins normally, leading to a motor neuron stress response. In particular, reduction in SMN causes a reduction in the levels of a chaperone protein that, Rubin said, acts like a protein traffic guard inside motor neurons. When other proteins are not folding correctly, the chaperone will hold up the stop sign, giving the other protein time to fold correctly before moving through the rest of the cell. If there are too few functional chaperones, it causes a protein traffic jam inside the cell. The buildup of these other proteins activates a stress response.
When the research team shut off the stress response in the SMA-affected cells, both in culture and in a mouse model, they were able to keep the motor neuron cells from dying. “You could say that ALS and SMA converge on a pathway that causes motor neurons to be stressed,” Rubin said.
Even though the two diseases have different origins — one with too much of the disease protein in the cells and the other with too little — the point of convergence may be important, said Rubin. Because they both involve a stress response in motor neurons specifically, it is possible that the two diseases could eventually be treated by one drug.





